
Gene Therapy
What is Gene Therapy?
Certain diseases are caused by faulty genes which produce defective proteins. The symptoms of genetic disease are the result of subsequent disrupted vital cell processes caused by missing or defective proteins. In the Bio Building Blocks section of this web-site, protein synthesis is outlined as the process whereby, genes ultimately give rise to proteins which are responsible for important cell processes. If a particular gene is defective, its protein product may not be made at all, may work poorly or may behave too aggressively.
For example: Cystic Fibrosis (CF) is caused by a missing or mutated gene that results in a defective cell membrane transport protein. This ultimately results in a build-up of thick mucus in the lungs and the body's airways. As another example, cancers are caused by cells that divide and grow uncontrollably. Particular genes can cause such cell growth to occur if they are defective. Such defective genes are called oncogenes.
Are we treating the symptom or treating the cause? Historically, genetic disorders have been treated by addressing the biological events that result from the genetic mutation, as opposed to fixing a defective gene (or genes) the ultimate source of the problem. For example, the treatment of diabetes has historically involved the administration of insulin (a protein), instead of fixing the defective genes in pancreatic cells that actually prevent these cells from producing insulin in the proper amounts, on their own.
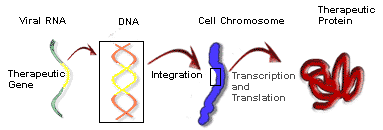
Gene therapy is an alternative approach whereby a genetic disorder is treated by inserting or integrating new genes into human cells. Many attempts at gene therapy aim to add a useful gene into a selected cell type to compensate for a missing or defective version. Other efforts aim to instill new properties in the target cell. This latter method is often employed in the treatment of cancer, where toxic genes are added to cancer cells in an effort to eliminate them. For an overview of how a specific gene is located and isolated from its source (so that it can be introduced into the patient) see our Genetic Engineering section .
It should be noted that even the most advanced somatic cell therapy techniques are still in clinical trials, and are not yet approved for general application. Much more research is required to develop safe, reliable gene therapy techniques.
Depending on the cell types affected, gene therapy can be classified into two broad categories: germ-line gene therapy and somatic cell gene therapy. Germ-line therapy occurs when germ cells (reproductive cells) are altered, meaning that the resulting genetic changes will be passed on to the patient's offspring. Alternatively, somatic cell gene therapy involves the alteration of somatic cells (non-reproductive body cells, like skin, brain or muscle cells). This genetic manipulation will only affect the individual to which the changes were made. Somatic cell gene therapy is the only type presently being considered in humans.
HOW ARE GENES INSERTED INTO TARGET CELLS?
Suppose a patient is afflicted with a genetic disorder that affected only certain cells in her or his brain. How could she or he be treated using gene therapy so that the therapeutic gene targets only those cells affected by the disorder? One solution is through the use of a vector. A vector is simply a "transporter" for the genetic material that allows it to enter the target cell and, depending on the vector type, can cause new genes to be integrated into the host cell genome. Vectors must be administered to target specific cell types.
There are three principal ways in which vectors can be administered to carry new genes into target cells. The first is called ex vivosomatic gene therapy, where the target cells are removed from the body, cultured in the laboratory with a vector, and re-inserted into the body. This process is usually carried out using blood cells because they are the easiest to remove and return.
The second option, in situ somatic gene therapy, occurs when the vector is placed directly into the affected tissue. This process is being developed for the treatment of cystic fibrosis (by direct infusion of the vector into the bronchi of the lungs), to destroy tumours (eg: brain cancer), and for the treatment of muscular dystrophy.
The third option is in vivo somatic gene therapy, where the vector is injected into the bloodstream , and is able to find and insert new genes only into the cells for which it was specifically designed. Although there are presently no in vivo treatments available, a breakthrough in this area will make gene therapy a very attractive option for treatment.In this case the vector designed to treat our hypothetical patient could be injected into a blood vessel in her or his arm and would find its way to the affected brain cells!
Vectors used in gene therapy can be classified as either viral or non-viral.
Viral Vectors
Both DNA and RNA viruses are being developed as vectors for use in gene therapy. Viruses are an excellent choice for use as vectors, because they have gained, through long periods of evolution, the ability to avoid destruction by the human immune system, and the capacity to get their own genetic material inside human cells. As discussed in the Bio Building Blocks section , viruses consist of genetic material (DNA or RNA) surrounded by a protective coat made of proteins and occasionally other molecule types as well.
Normally, a virus infects a cell when its genetic material enters it. Once the viral genetic material is inside, it "hijacks" the cell's DNA- and protein-making machinery, causing it to produce new viruses. Some viruses are even capable of integrating their own genetic material into the host cell's genome.
It is the outer protective viral coat that allows the inner genetic material to penetrate the cell. This outer coat also determines the type of cell that a given virus will infect. Once inside, it is the harmful viral genes that actually hijack the cell and eventually cause it to die.
To trick the virus, scientists retain the outer viral coat, but modify the inner genetic material. They remove the harmful genes and replace them with therapeutic ones. Now the virus is pathogenically disabled (it is no longer harmful to the cell it infects) and incapable of reproducing itself. However, it retains its capability to transfer its genetic material to the cells for which its outer coat was designed. The transfer of genetic material by way of a viral vector is called transduction.
Retroviral Vectors
The structure and mode of infection of retroviruses is discussed in the Bio Building Blocks section. Briefly, retroviruses have RNA as their genetic material. These viruses also carry a special enzyme that, once inside a cell, makes double-stranded DNA from the virus' RNA template. The new DNA becomes incorporated into the host cell's genome. When the "new" chromosomal genes are transcribed, new virus particles are made, which will leave the cell to infect other cells.
Most types of retroviruses are not very harmful to the cell. Even though all viruses to be used as vectors are deactivated,' meaning that their harmful genes are removed, the fact that the types of retroviruses presently being used as vectors are not very harmful in their natural forms means that their use poses less risk than the use of some other viruses. Even if something goes wrong and some of the original retrovirus particles are administered to the patient, they will not cause serious problems.
The murine leukaemia virus (MuLV) is one of the more popular retroviruses used as a retroviral vector. The reproductive genes in the retrovirus are replaced with the therapeutic gene. When the virus infects the cell, the therapeutic gene gets incorporated into the cell chromosomes. The new gene causes a protein to be produced which is hoped to have some positive therapeutic effects, either providing an otherwise missing protein, or causing the destruction of harmful cells.
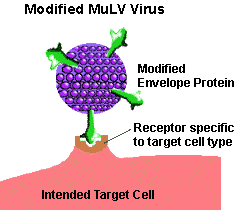
There are several challenges that scientists must overcome for effective in vivo treatment of disease using retroviral vectors. For example, the viruses must be capable of targeting only those cells affected by the disorder. If this were the case, they could be injected directly into the bloodstream (in vivo gene therapy) where they would become dispersed throughout the body, but would only transduce those cells for which they were designed. Presently, retroviral vectors are not terribly specific, meaning that many cells not intended for the transfer of the gene are transduced by the virus, which reduces the transfer to the targeted cell population.
To understand how viruses can be made to be more specific, we should consider how viruses "choose" the cells they infect. A virus must bind to specific surface receptor molecules to gain entry into a cell. To this end, retroviruses have outer envelope proteins that fit perfectly into certain receptors on specific cells. The MuLV virus binds to cells containing a receptor called the amphotropic receptor. The problem is that a broad range of cell types possess the amphotropic receptor. This means that the MuLV virus, in its natural form, can infect all of these cell types, most of which are likely not the target of the therapy!
To make retroviral vectors more specific about the cells they invade, scientists are experimenting with ways of replacing or modifying the outer viral proteins, so that they fit into more rare receptors that appear only on specific cell types being targeted for therapy. Another approach has been to add new proteins to the outer viral envelope which either better recognize the target cell, or better recognize the region of the body where the target cells are located.
Another challenge is to engineer retroviral vectors to transduce non-dividing cells. Most retroviruses target actively dividing cells, which makes them ideal for the treatment of rapidly dividing tumour cells, but not in situations where a therapeutic gene is to be introduced into a non-dividing cell, like in the treatment of cystic fibrosis mentioned above. Those few retroviruses that have the ability to infect non-dividing cells are the harmful ones (HIV, the virus that results in AIDS, is one of them). HIV viruses (with their harmful genes removed) cannot be used as vectors, because even with the removal of these genes, there is still a possibility that the virus might become harmful again through a process called recombination. To virtually eliminate the possibility that harmful viruses are produced in this way, while still harnessing the capability of HIV to transduce non-dividing cells, scientists are experimenting with the development of hybrid vectors, made up mostly of other retroviruses and which contain very small and harmless parts of the HIV virus.
As of April, 1998, there was only one vector-based therapeutic technique in the final clinical trial stage (called Phase III). This technique employs a retroviral vector called G1TkSvNa for the treatment of glioblastoma multiforma, a malignant brain tumour. The treatment is an in situ therapeutic technique, where mouse cells capable of producing and secreting the vector are injected into the tumour. The secreted vectors infect only those cells that are rapidly dividing, meaning only the tumour cells and the vessels supplying blood to the tumour are transduced. The gene transduced into the tumour cells gives rise to a protein (called Herpes Simplex Thymidine Kinase or HSTk).Fourteen days later, a drug called ganciclovir is injected into the patient, which is toxic to any cell that incorporates it into its DNA. Only the cells containing HSTk (the tumour cells) are capable of incorporating ganciclovir into their DNA and these cells are therefore selectively killed off.
Adenoviral Vectors
Adenoviruses are DNA viruses that are able to transduce a large number of cell types, including non-dividing cells. Adenoviruses also have the capacity to carry long segments of added genetic information. In addition, it is fairly easy to produce large amounts of adenoviruses in culture. Adenoviruses, in their natural form, are not very harmful, typically causing nothing more serious than a chest cold in otherwise healthy people. This means that their use as vectors is quite safe. For all these reasons, adenoviruses are currently the most widely used DNA vectors for experiments in in situ gene therapy. Research is currently under way using adenoviral vectors for the treatment of several cancers and cystic fibrosis.
The size of the adenovirus protein coat is just large enough to fit the original viral DNA inside. As a result, for every new therapeutic gene to be inserted into the viral genome, a corresponding piece of the old viral DNA must be removed. To make room for the new therapeutic DNA, a region of the old viral DNA called E3 is sometimes removed. However, removing the E3 region has drawbacks, because it codes for a protein that suppresses the human immune response against the vector. Without the E3 region, the virus is more susceptible to the immune system and is more likely to be destroyed before it has served its purpose.
Adenoviral vectors send their DNA to the nucleus, but the DNA does not get incorporated into the host cell's chromosomes. For this reason, the viral DNA has a finite lifetime within the cell before it is degraded, meaning that the added genes are effective only temporarily. Treatments for chronic conditions like cystic fibrosis, therefore, would need to be repeated periodically, perhaps on a monthly or yearly basis. On the other hand, the transient nature of therapeutic gene expression is useful when the added genes are needed temporarily to induce an immune response to a cancer or pathogen.
Other Viral Vectors
Among the other virus types being explored as vectors are the adeno-associated virus (AAV) and the herpes simplex virus (HSV). Both are DNA-based viruses. AAV integrates its genetic material into a host chromosome and cause no diseases in humans. However, because AAV are small, they cannot accommodate large genes. HSV vectors do not integrate their genes into the host genome. They tend to target neurons and thus have the potential for use in the treatment of neurological disorders.
Non-Viral Vectors
The use of non-viral vectors can involve a direct injection of plasmid DNA or mixing plasmid DNA with compounds that allow it to cross the cell membrane and protect the DNA from degradation. These methods are currently less efficient than the use of viral vectors. However, unlike disabled viruses which have the possibility of changing spontaneously and causing disease, non-viral vectors possess no viral genes and therefore cannot cause disease.
Using Liposomes
Liposomes are small, hollow spheres of fatty molecules that are capable of carrying DNA inside of them. A liposome can fuse with the cell membrane, releasing its contents into the cell interior.
Plasmid DNA containing the therapeutic gene is incubated with the empty liposomes under specific conditions. The negatively charged DNA binds to the positively charged (called cationic) liposomes and the plasmids are absorbed. Liposomes containing plasmid DNA are called lipoplexes. The lipoplexes can subsequently enter the cells of interest, and thus introduce the therapeutic DNA into the cells.
Experiments have been carried out where lipoplexes have been injected into tumours. The lipolexes contained a gene that gives rise to a protein that is recognized by the human immune system. Theoretically, these genes should cause the tumour cells to express the recognizable protein on their surface, which will mark the cells for destruction by the immune system.
The use of lipoplexes for the treatment of cystic fibrosis is currently being studied as well. The cause of the illness is a defective gene which causes a particular protein in the patient's lung cells to be defective. The lipoplexes that are administered using an aerosol spray into the patient's lungs, contain the gene for a functional version of the protein.
Lipoplexes are not as efficient as viral vectors in introducing genes into cells. To improve their efficiency, scientists are attempting to incorporate some viral proteins into the outer surfaces of lipoplexes. In particular, the viral proteins that recognize and bind to specific molecules on the host cell's surface, are being incorporated.
Injecting Plasmid DNA
Muscle cells have been shown to be capable of taking up and expressing plasmid DNA. This raises the possibility that plasmid DNA injected into muscles could stimulate the production by muscle cells of a therapeutic protein. This protein could then be secreted into the bloodstream and to the rest of the body. For example, the gene coding for erythropoietin (a protein which helps stimulate the production of red blood cells) has been experimentally injected into animal muscles with some success. Such a treatment would be useful to patients after chemotherapy or radiation therapy.
In addition, plasmid DNA shows promise for use in vaccines, stimulating protective immune responses against diseases like herpes, AIDS or malaria. When the plasmid DNA is injected into muscles, it enters muscle cells and as a result, causes the cells to produce the proteins that correspond to the genes the plasmids contain. The immune system will then learn to recognize the new proteins and will destroy them if they are encountered in the future. Experiments are currently under way where plasmids containing genes for viral coat proteins are injected, in attempt to make the immune system recognize these viruses, so that it will attack and destroy them if they are ever encountered.
"ANTISENSE" TECHNOLOGY
As discussed in the Bio Building Blocks section , viruses hijack cellular machinery to produce their own proteins and to replicate their genetic material, which results in the production of new viruses. One of the potential uses of antisense technology is to prevent viruses that infect a host cell from producing their own proteins. This would, in turn, prevent their replication.
Recall that proteins are constructed through a two step process . In the first step, DNA is transcribed to produce messenger RNA (mRNA) . The second step involves thetranslation of the mRNA to make a protein. Antisense drugs interact with mRNA, preventing them from being translated into their corresponding protein.
An mRNA molecule is a chain of nucleotides, that gets "read" by a ribosome in the synthesis of a protein. An antisense drug is an oligonucleotide (a relatively small, single stranded chain of nucleotides) that is complementary to a small segment of a target mRNA molecule. When the drug comes into contact with its complementary mRNA, it binds to the mRNA in the same way as the two strands of a DNA molecule bind together. This makes the mRNA "unreadable" by the ribosome, and so no protein is produced.
Because an antisense drug is designed to be complementary to a particular mRNA sequence that is specific to a particular virus' mRNA, it will not interfere with any of the host cell's naturally produced mRNA, meaning that the side effects of the drug are minimal.
At the end of August, 1998, the US Food and Drug Administration (FDA) approved a drug called formivirsen for the treatment of cytomegalovirus (CMV) retinitis in patients with AIDS. This makes formivirsen the first antisense drug on the market.Formivirsen blocks the replication of cytomegalovirus (CMV) which causes retinitis, an eye infection leading to blindness that mainly affects AIDS patients. The drug is periodically injected into the patient's eye, and is claimed to cause only mild side-effects as compared to some other antiviral drugs.